
![]()
Written by Seán Kavanagh, along with contributions from group members (and alumni) Alex Squires, Adair Nicolson, Irea Mosquera-Lois, Alex Ganose, and Bonan Zhu, doped is a Python package for managing solid-state defect calculations, with functionality to generate defect structures and relevant competing phases (for chemical potentials), interface with
ShakeNBreak for defect structure-searching (see below), write VASP input files for defect supercell calculations, and automatically parse and analyse the results.
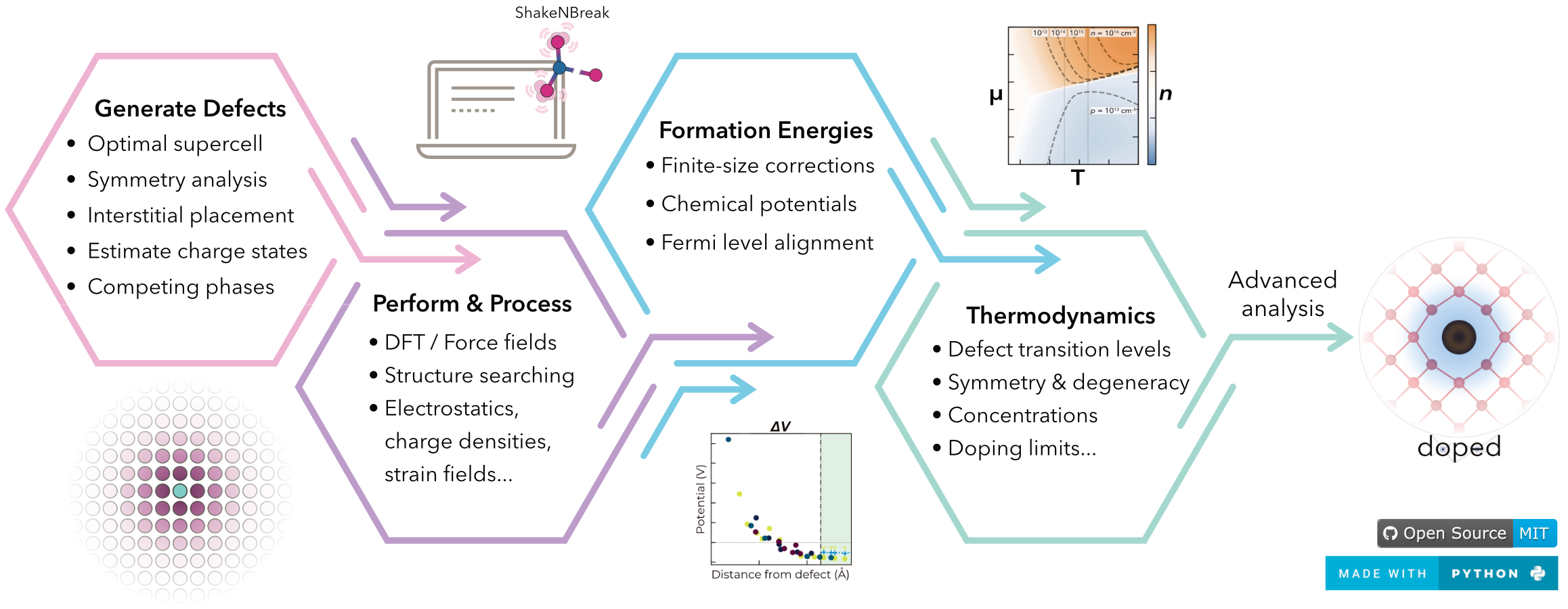
Tutorials showing the code functionality and usage are provided on the doped documentation site.
If you use doped in your work, please cite the JOSS paper:
S. R. Kavanagh et al. doped: Python toolkit for robust and repeatable charged defect supercell calculations, Journal of Open Source Software 9 (96), 6433, 2024
Publications using doped
See the updated list on the doped documentation site!
- X. Wang et al. Upper efficiency limit of Sb2Se3 solar cells arXiv 2024
- I. Mosquera-Lois et al. Machine-learning structural reconstructions for accelerated point defect calculations arXiv 2024
- W. Dou et al. Giant Band Degeneracy via Orbital Engineering Enhances Thermoelectric Performance from Sb2Si2Te6 to Sc2Si2Te6 ChemRxiv 2024
- K. Li et al. Computational Prediction of an Antimony-based n-type Transparent Conducting Oxide: F-doped Sb2O5 Chemistry of Materials 2023
- X. Wang et al. Four-electron negative-U vacancy defects in antimony selenide Physical Review B 2023
- Y. Kumagai et al. Alkali Mono-Pnictides: A New Class of Photovoltaic Materials by Element Mutation PRX Energy 2023
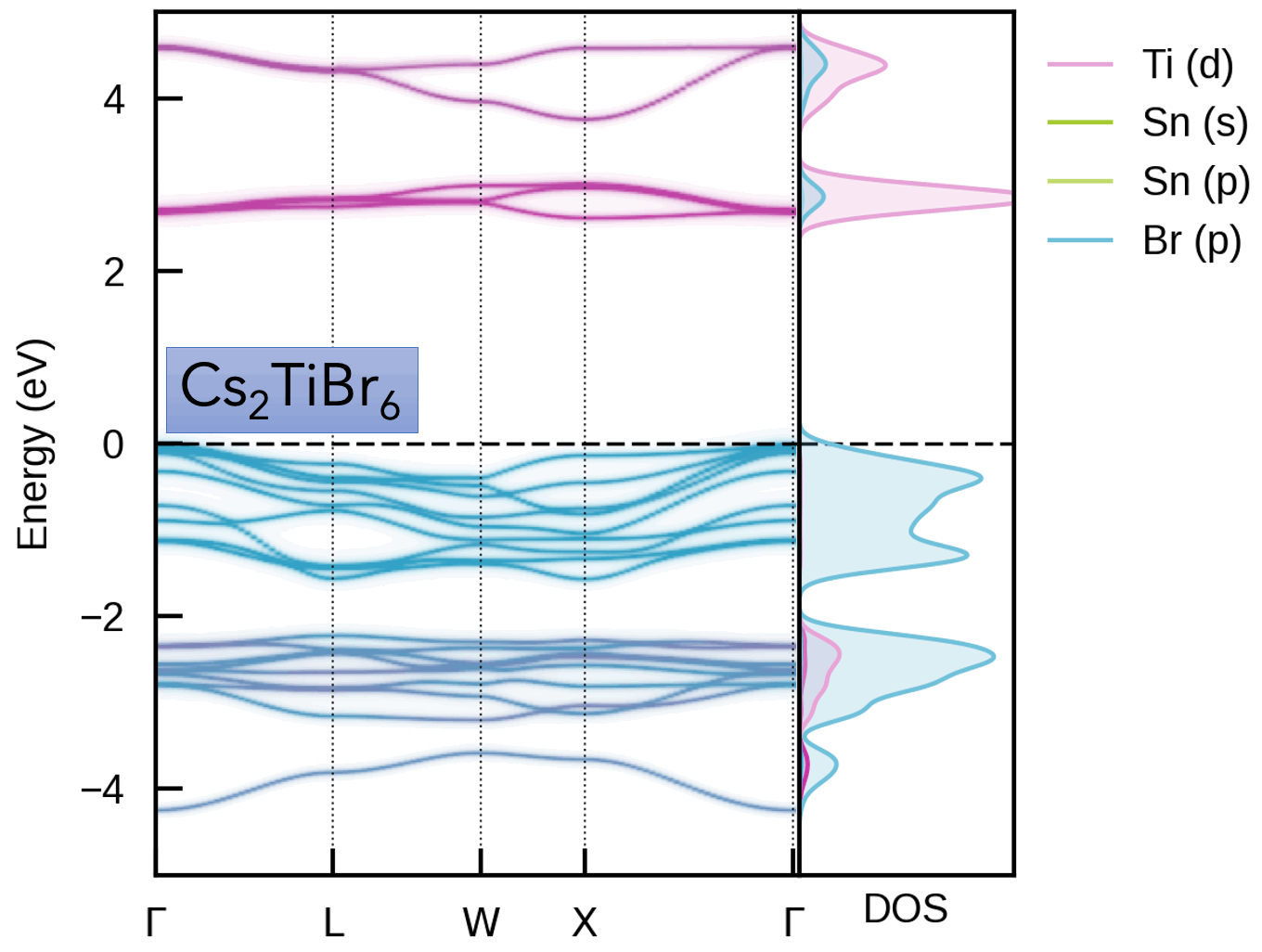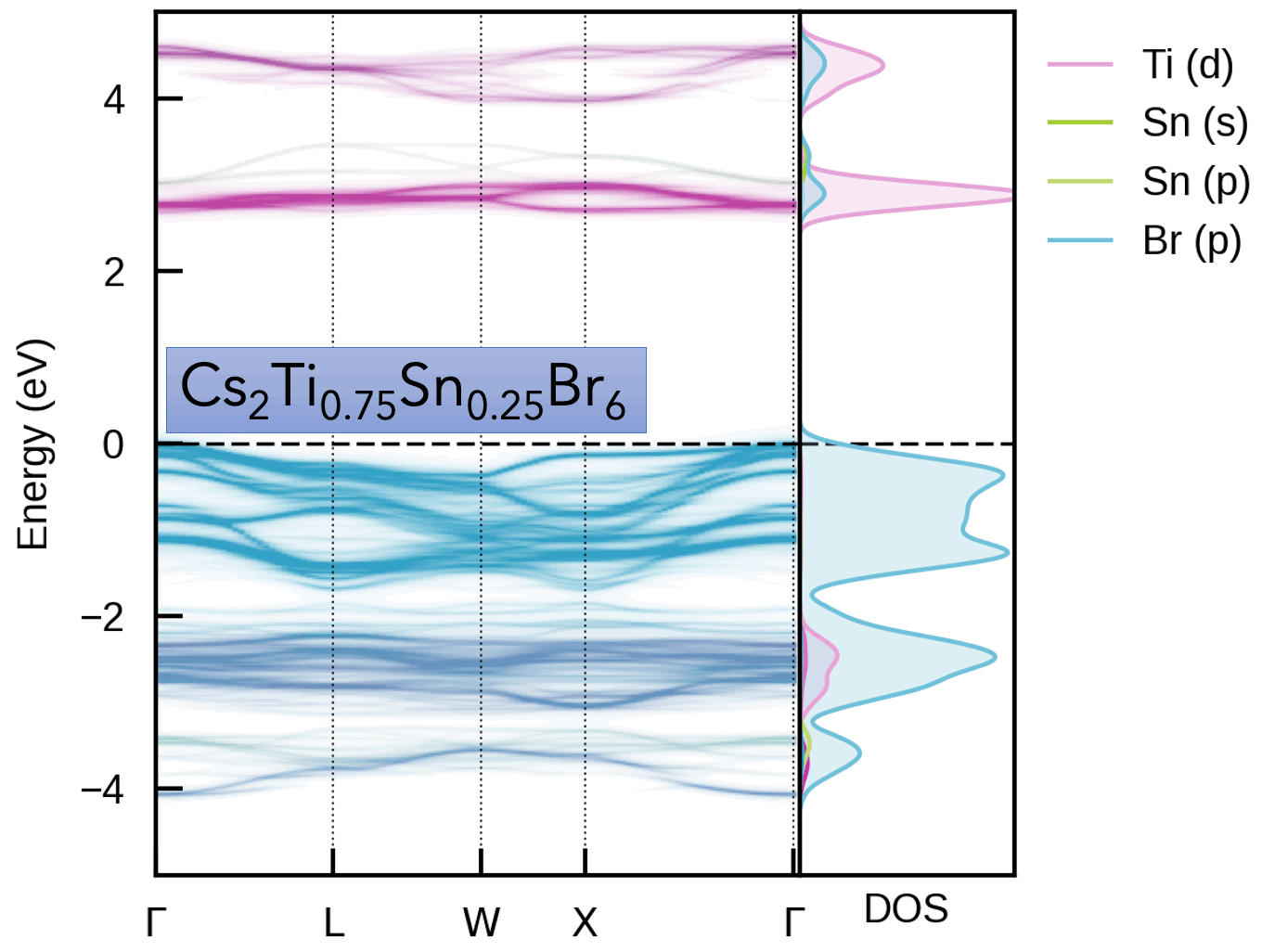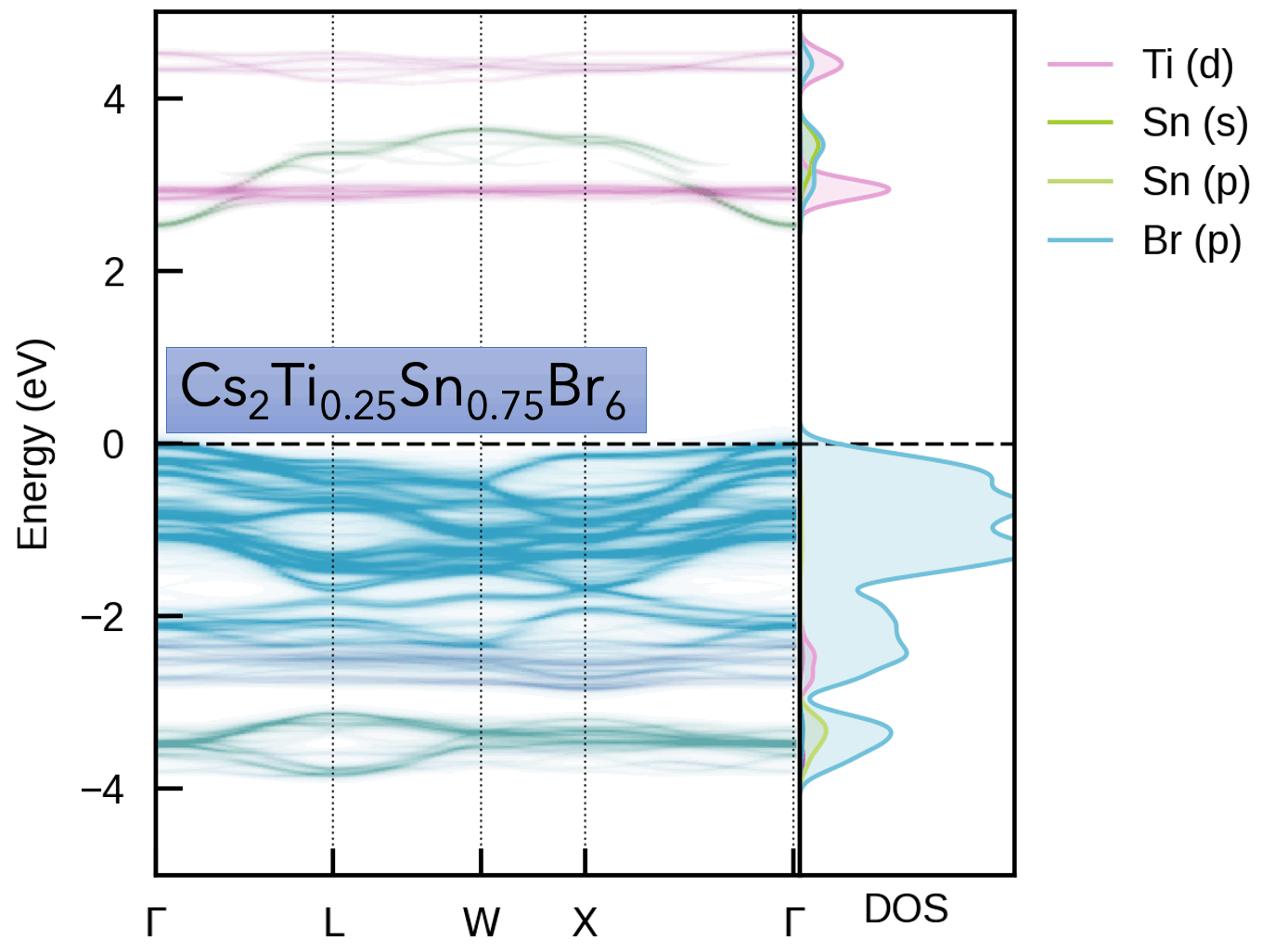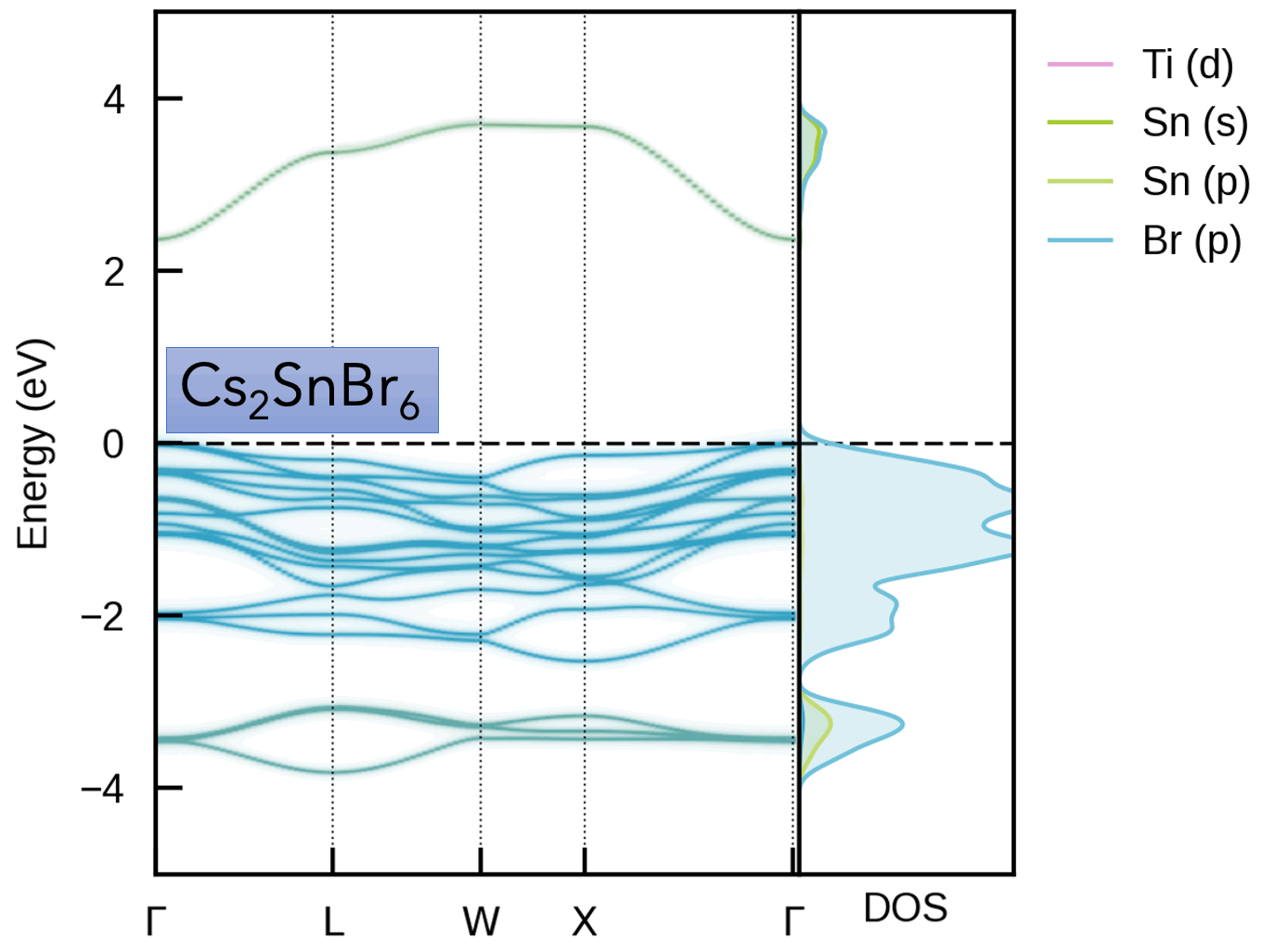
- S. M. Liga & S. R. Kavanagh, A. Walsh, D. O. Scanlon, G. Konstantatos Mixed-Cation Vacancy-Ordered Perovskites (Cs2Ti1–xSnxX6; X = I or Br): Low-Temperature Miscibility, Additivity, and Tunable Stability Journal of Physical Chemistry C 2023
- A. T. J. Nicolson et al. Cu2SiSe3 as a promising solar absorber: harnessing cation dissimilarity to avoid killer antisites Journal of Materials Chemistry A 2023
- Y. W. Woo, Z. Li, Y-K. Jung, J-S. Park, A. Walsh Inhomogeneous Defect Distribution in Mixed-Polytype Metal Halide Perovskites ACS Energy Letters 2023
- P. A. Hyde et al. Lithium Intercalation into the Excitonic Insulator Candidate Ta2NiSe5 Inorganic Chemistry 2023
- J. Willis, K. B. Spooner, D. O. Scanlon On the possibility of p-type doping in barium stannate Applied Physics Letters 2023
- J. Cen et al. Cation disorder dominates the defect chemistry of high-voltage LiMn1.5Ni0.5O4 (LMNO) spinel cathodes Journal of Materials Chemistry A 2023
- J. Willis & R. Claes et al. Limits to Hole Mobility and Doping in Copper Iodide Chemistry of Materials 2023
- I. Mosquera-Lois & S. R. Kavanagh, A. Walsh, D. O. Scanlon Identifying the ground state structures of point defects in solids npj Computational Materials 2023
- Y. T. Huang & S. R. Kavanagh et al. Strong absorption and ultrafast localisation in NaBiS2 nanocrystals with slow charge-carrier recombination Nature Communications 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh, C. Freysoldt Impact of metastable defect structures on carrier recombination in solar cells Faraday Discussions 2022
- Y-S. Choi et al. Intrinsic Defects and Their Role in the Phase Transition of Na-Ion Anode Na2Ti3O7 ACS Applied Energy Materials 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh Rapid Recombination by Cadmium Vacancies in CdTe ACS Energy Letters 2021
- C. J. Krajewska et al. Enhanced visible light absorption in layered Cs3Bi2Br9 through mixed-valence Sn(II)/Sn(IV) doping Chemical Science 2021
- Dou W., Spooner K. B., Kavanagh S. R., Zhou M. & Scanlon, D. O. Band Degeneracy and Anisotropy Enhances Thermoelectric Performance from Sb2Si2Te6 to Sc2Si2Te6 Journal of the American Chemical Society 2024, 146(26), 17679.
- Hachmioune S., Ganose A. M., Sullivan M. B. & Scanlon, D. O. Exploring the Thermoelectric Potential of MgB4: Electronic Band Structure, Transport Properties, and Defect Chemistry Chemistry of Materials 2024, 36(12), 6062.
- Huo L. & Savory C. N. Assessing the Electronic and Optical Properties of Lanthanum Diselenide: a Computational Study Journal of Materials Chemistry C 2024
- Li K., Willis J., Kavanagh S. R. & Scanlon, D. O. Computational Prediction of an Antimony-Based n-Type Transparent Conducting Oxide: F-Doped Sb2O5 Chemistry of Materials 2024, 36(6), 2907.
- Yeganeh M. & Fakhrabad D. V. Lattice Thermal Conductivity and Thermoelectric Properties of Two-Dimensional Honeycomb Monolayer of CdO Solid State Communications 2024, 378, 115391.
- Han D., Zhu B., Cai Z., Spooner K. B., Rudel S. S., Schnick W., Bein T., Scanlon, D. O. & Ebert H. Discovery of Multi-Anion Antiperovskites X6NFSn2 (X= Ca, Sr) as Promising Thermoelectric Materials by Computational Screening Matter. 2024, 7(1), 158.
- Fu Y., Lohan H., Righetto M., Huang Y. T., Kavanagh S. R., Cho C. W., Zelewski S. J., Woo Y. W., Demetriou H., McLachlan M. A., Heutz S., Piot B. A., Scanlon, D. O., Rao, A., Herz, L. M., Walsh, A. & Hoye, R. L. Z. Factors Enabling Delocalized Charge-Carriers in Pnictogen-Based Solar Absorbers: in-Depth Investigation into CuSbSe2. arXiv 2024.
- Willis J., Spooner K. B. & Scanlon, D. O. On the Possibility of p-Type Doping in Barium Stannate Applied Physics Letters. 2023, 123(16), 162103.
- Zeeshan M., Vishwakarma C. K. & Mani B. K. First-Principles Study of Disordered Half-Heusler Alloys XFe0.5Ni0.5Sn (X = Nb, Ta) as Thermoelectric Prospects arXiv 2023.
- Rodriguez L. H., Spooner K.B., Einhorn M. & Scanlon, D. O. Sr2Sb2O7: a Novel Earth Abundant Oxide Thermoelectric Journal of Materials Chemistry C 2023, 11(27), 9124.
- Brlec K., Spooner K. B., Skelton J. M. & Scanlon, D. O. Y2Ti2O5S2 — a Promising n-Type Oxysulphide for Thermoelectric Applications Journal of Materials Chemistry A. 2022, 10(32) 16813.
- Spooner K.B., Ganose A.M., Leung W.W., Buckeridge J., Williamson B.A., Palgrave R.G. & Scanlon, D. O. BaBi2O6: a Promising n-Type Thermoelectric Oxide with the PbSb2O6 Crystal Structure Chemistry of Materials 2021, 33(18), 7441.
- Kavanagh S. R., Savory C. N., Scanlon, D. O. & Walsh A. Hidden Spontaneous Polarisation in the Chalcohalide Photovoltaic Absorber Sn2SbS2I3 Materials Horizons 2021, 8(10), 2709.
- Spooner, K. B., Ganose, A. M. & Scanlon, D. O. Assessing the Limitations of Transparent Conducting Oxides as Thermoelectrics Journal of Materials Chemistry A 2020, 8(24), 11948.
![]()
Written by Kieran Spooner, along with contributions from alumni Maud Einhorn and Daniel Davies, ThermoParser is a Python package for analysing electronic and thermal transport properties, through unifying data from diverse codes and providing CLI and Python interfaces to simply and transparently retrieve data, calculate emergent properties and produce publication-ready visualisations.
Tutorials showing the code functionality and usage are provided on the ThermoParser documentation site.
If you use ThermoParser in your work, please cite the JOSS paper:
Spooner, K. B., Einhorn, M., Davies, D. W., & Scanlon, D. O. ThermoParser: Streamlined Analysis of Thermoelectric Properties Journal of Open Source Software 2024, 9(97), 6340.
Publications using ThermoParser
This list includes references under the previous name, ThermoPlotter.
Written by Bonan Zhu and Seán Kavanagh, easyunfold is a simple, easy-to-use, yet powerful and flexible tool which implements the band structure unfolding workflow using plane-wave DFT codes (such as VASP or CASTEP), all the way from input file generation to publication-quality plotting, as
well as carrier effective mass analysis.
Tutorials showing the code functionality and usage are provided on the easyunfold documentation site.
Publications using easyunfold
See the updated list on the easyunfold documentation site!
- S. M. Liga & S. R. Kavanagh, A. Walsh, D. O. Scanlon, G. Konstantatos Mixed-Cation Vacancy-Ordered Perovskites (Cs2Ti1–xSnxX6; X = I or Br): Low-Temperature Miscibility, Additivity, and Tunable Stability Journal of Physical Chemistry C 2023
- A. T. J. Nicolson et al. Interplay of Static and Dynamic Disorder in the Mixed-Metal Chalcohalide Sn2SbS2I3 Journal of the Americal Chemical Society 2023
- Y. T. Huang & S. R. Kavanagh et al. Strong absorption and ultrafast localisation in NaBiS2 nanocrystals with slow charge-carrier recombination Nature Communications 2022
- Y. Wang & S. R. Kavanagh et al. Cation disorder engineering yields AgBiS2 nanocrystals with enhanced optical absorption for efficient ultrathin solar cells Nature Photonics 2022 (Early version)
Written by Seán Kavanagh and
Irea Mosquera-Lois, ShakeNBreak (“SnB”) implements the defect structure-searching approach detailed in ‘Identifying the Ground State Structures of Defects in Solids‘, npj Comput Mater 9, 25, 2023. This method employing chemically-guided bond distortions to locate ground-state and metastable structures of point defects in solid materials.
Main features include:
- Defect structure generation:
- Automatic generation of distorted structures for input defects
- Optionally, input file generation for geometry optimisation with several codes (VASP, CP2K,
Quantum-Espresso, CASTEP & FHI-aims)
- Defect structure analysis:
- Automatic parsing of geometry relaxation results
- Plotting of final energies versus distortion to demonstrate what energy-lowering reconstructions have been identified
- Coordination & bonding analysis to investigate the physico-chemical factors driving an energy-lowering distortion
- Magnetisation analysis (currently only supported for VASP)

The code supports several DFT codes, including VASP, CP2K, Quantum-Espresso, CASTEP and FHI-aims.
If you use ShakeNBreak in your work, please cite the following:
- Code: Mosquera-Lois, I. & Kavanagh, S. R.; Walsh, A.; Scanlon, D. O. ShakeNBreak: Navigating the defect configurational landscape, Journal of Open Source Software 7 (80), 4817, 2022
- Theory/Method: Mosquera-Lois, I. & Kavanagh, S. R.; Walsh, A.; Scanlon, D. O. Identifying the Ground State Structures of Defects in Solids, npj Comput Mater 9, 25, 2023
You may also find the following literature useful:
- Preview: Mosquera-Lois, I.; Kavanagh, S. R. In Search of Hidden Defects, Matter 4 (8), 2602-2605, 2021
- News & Views: Mannodi-Kanakkithodi, A. The Devil is in the Defects, Nature Physics 2023 (Free-to-read link)
Publications using ShakeNBreak
See the updated list on the ShakeNBreak documentation site!
- X. Wang et al. Upper efficiency limit of Sb2Se3 solar cells arXiv 2024
- I. Mosquera-Lois et al. Machine-learning structural reconstructions for accelerated point defect calculations arXiv 2024
- K. Li et al. Computational Prediction of an Antimony-based n-type Transparent Conducting Oxide: F-doped Sb2O5 Chemistry of Materials 2024
- X. Wang et al. Four-electron negative-U vacancy defects in antimony selenide Physical Review B 2023
- Y. Kumagai et al. Alkali Mono-Pnictides: A New Class of Photovoltaic Materials by Element Mutation PRX Energy 2023
- A. T. J. Nicolson et al. Cu2SiSe3 as a promising solar absorber: harnessing cation dissimilarity to avoid killer antisites Journal of Materials Chemistry A 2023
- J. Willis, K. B. Spooner, D. O. Scanlon On the possibility of p-type doping in barium stannate Applied Physics Letters 2023
- J. Cen et al. Cation disorder dominates the defect chemistry of high-voltage LiMn1.5Ni0.5O4 (LMNO) spinel cathodes Journal of Materials Chemistry A 2023
- J. Willis & R. Claes et al. Limits to Hole Mobility and Doping in Copper Iodide Chemistry of Materials 2023
- I. Mosquera-Lois & S. R. Kavanagh, A. Walsh, D. O. Scanlon Identifying the ground state structures of point defects in solids npj Computational Materials 2023
- B. Peng et al. Advancing understanding of structural, electronic, and magnetic properties in 3d-transition-metal TM-doped α-Ga₂O₃ (TM = V, Cr, Mn, and Fe) Journal of Applied Physics 2023
- Y. T. Huang & S. R. Kavanagh et al. Strong absorption and ultrafast localisation in NaBiS2 nanocrystals with slow charge-carrier recombination Nature Communications 2022
- S. R. Kavanagh, D. O. Scanlon, A. Walsh, C. Freysoldt Impact of metastable defect structures on carrier recombination in solar cells Faraday Discussions 2022
- Y-S. Choi et al. Intrinsic Defects and Their Role in the Phase Transition of Na-Ion Anode Na2Ti3O7 ACS Applied Energy Materials 2022 (Early version)
- S. R. Kavanagh, D. O. Scanlon, A. Walsh Rapid Recombination by Cadmium Vacancies in CdTe ACS Energy Letters 2021
- C. J. Krajewska et al. Enhanced visible light absorption in layered Cs3Bi2Br9 through mixed-valence Sn(II)/Sn(IV) doping Chemical Science 2021 (Early version)
- (News & Views): A. Mannodi-Kanakkithodi The devil is in the defects Nature Physics 2023 (Free-to-read link)
Written by Dr Alex Squires and Dr Ben Morgan, py-sc-fermi is a Python package for calculating the concentration of point defects in (semiconducting) crystalline materials. The required inputs are the volume, density of states of the bulk material, and the formation energies and degeneracies of the point defects. The outputs include the self consistent Fermi energy, defect transition levels, and concentrations of the point defects, electrons and holes at a given temperature. py-sc-fermi uses a numerical method to solve for the self-consistent Fermi level in a material, necessary for accurately quantifying the populations of point defects in such materials.

If you use py-sc-fermi in your work, please consider cite the following:
- the paper associated with the py-sc-fermi
- the paper associated with the FORTRAN code SC-Fermi on which this core algorithm was initially based, which provides an excellent discussion of the underlying theory.
Written by Katarina Brlec and Daniel Davies, surfaxe (click here to get the source code) is a python package for automating and simplifying density functional theory (DFT) calculations of surface properties, as well as providing analytical tools for bulk and surface calculations. The code makes extensive use of pymatgen surface modules with full functionality retained. Full integration with FireWorks and AiiDA is possible for managing calculations on high-performance clusters. As well as a fully flexible python API, surfaxe has a lightweight command line interface. surfaxe primarily supports VASP, however the slab generation module is code-agnostic. Support for other DFT codes is planned for future releases.
The modularity of surfaxe follows a best-practice workflow for the calculation of surface properties. Generation module contains scripts for automatic cleaving of slabs from the bulk and organising them into a directory structure for convergence testing with respect to slab and vacuum thickness. Convergence and analysis modules allow for analysis of atomic displacements, bond lengths, electrostatic potential and energies of slabs. Data module wraps up final collation of data, with automated extraction of surface energy, vacuum and core energy levels, along with the necessary calculation parameters.

If you do use surfaxe, please cite the following paper in your publication:
K. Brlec, D. W. Davies and D. O. Scanlon, Surfaxe: Systematic surface calculations. Journal of Open Source Software, 6(61), 3171, (2021) DOI: 10.21105/joss.03171
Written by Alex Ganose and Dr Adam Jackson, sumo is a Python package for plotting and analysis of materials chemistry ab initio calculation data. sumo (click here to get the source code) is a set of command-line tools for publication-ready plotting and analysis of ab initio calculation data. The code includes a fully-documented Python module, upon which the command-line scripts are built. sumo currently only supports VASP, however, extending the code to other ab initio calculators is planned for future releases. The code relies on several open-source Python packages for common tasks, including pymatgen for data loading, spglib for symmetry analysis, and Matplotlib for plotting.
The main plotting functionality of sumo includes density of states plots, electronic and phonon band structure diagrams, and optical absorption spectra (as shown in the Figure below). The code has been designed to allow for significant customisation of plots, including the ability to produce projected density of states and orbital resolved band structures. The code additionally supplies a tool for generating k-point paths along high-symmetry directions in the Brillouin zone, with the ability to write the necessary input files required to perform the calculations in VASP. Crucially, this tool allows a single band structure plot to be split into several ab initio calculations, as is essential when dealing with large materials or restrictive batch systems. Lastly, a script is provided to extract information from semiconductor band structures, including direct and indirect band gaps, band edge locations, and parabolic and non-parabolic effective masses.

If you do use SUMO, please cite the following paper in your publication:
A.M. Ganose, A. J. Jackson and D. O. Scanlon, sumo: Command-line tools for plotting and analysis of periodic ab initio calculations, Journal of Open Source Software, 3(28), 717 (2018) DOI: 10.21105/joss.00717
Publications using SUMO
-
- I. W. H. Oswald, E. M. Mozur, I. P. Mosely, H. Ahn and J. R. Neilson, Hybrid Charge-Transfer Semiconductors: (C7H7)SbI4, (C7H7)BiI4, and Their Halide Congeners, Inorganic Chemistry , 58, 5818 (2019) doi: 10.1021/acs.inorgchem.9b00170
-
- D. H. Cao, P. Guo, A Mannodi-Kanakkithodi, G. P. Wierderrecht, D. J. Gosztola, R. D. Schaller, M. K. Y. Chan and A. B. F. Martinson, Charge Transfer Dynamics of Phase-Segregated Halide Perovskites: CH3NH3PbCl3 and CH3NH3PbI3 or (C4H9NH3)2(CH3NH3)n−1PbnI3n+1 Mixtures, ACS Applied Materials & Interfaces, 11, 9583 (2019) doi: 10.1021/acsami.8b20928
-
- B. A. D. Williamson, G. J. Limburn, G. W. Watson, G. Hyett, and D. O. Scanlon, Computationally Driven Discovery of Layered Quinary Oxychalcogenides: Potential p-Type Transparent Conductors?, ? , Submitted (2019) ChemRxiv
-
- Z. Wang, A. M. Ganose, C. Niu, and D. O. Scanlon, Two-dimensional hybrid perovskites for tunable energy level alignments and photovoltaics, Journal of Materials Chemistry C, 7, 5139 (2019) doi: 10.1039/C9TC01325C
-
- J. T. Pegg, A. E. Shields, M. T. Storr, D. O. Scanlon, and N. H. de Leeuw, Noncollinear Relativistic DFT+U Calculations of Actinide Dioxide Surfaces, Journal of Physical Chemistry C, 123, 356 (2019) doi: 10.1021/acs.jpcc.8b07823
-
- W. W. W. Leung, C. N. Savory, R. G. Palgrave, and D. O. Scanlon, An experimental and theoretical study into NaSbS2 as an emerging solar absorber, Journal of Materials Chemistry C, 7, 2059 (2019) doi: 10.1039/C8TC06284F
Written by Dr Adam Jackson and Alex Ganose, GALORE simplifies and automates the process of simulating photoelectron spectra from ab initio calculations. This replaces the tedious process of extracting and interpolating crosssectional weights from reference data and generates tabulated data or publication-ready plots as needed. The broadening tools may also be used to obtain realistic simulated spectra from a theoretical set of discrete lines (e.g. infrared or Raman spectroscopy). GALORE is a Materials Design aid, as it can quickly convert calculated data to simulated spectra which can be compared easily with experiment.
GALORE (click here to get the source code) provides a command-line tool and Python API to import data and resample it to a dense, regular X-Y series. This mesh can then be convolved with Gaussian and Lorentzian functions to yield a smooth output, in the form of a plot or data file. Numpy functions are used for data manipulation and convolution on a finite grid and Matplotlib is used for plotting. As well as simple tabular data files, the electronic DOS or PDOS may be imported directly from the output of the VASP or GPAW codes. An example of the GALORE proceedure for generating simulated PES spectra is shown in the Figure below.

Cross-sectional weights are included for some standard energy values (He(II) UPS and Al k-alpha) from tabulated ab initio calculations. Users may provide their own weighting values in the same human-readable JSON file format. Higher-energy (HAXPES) spectra may be simulated using cross-sections from fitted data over an energy range 1-1500 keV. Tabulated data was fitted to an order-8 polynomial on a log-log scale, and coefficients for each element and orbital shape are stored in a database file. The fitting error is generally below 1%, with outliers in the region of 2–3%, as demonstrated in the Figure below. The order-8 fit was selected based on cross-validation in order to avoid over-fitting

Additional cross-sectional weights across a wider energy range and photoelectron angular distribution parameters (from ADNDT calculations here and here) have been mined and digitised by Joe Willis et al., with the original authors’ permission. These data allow for accurate cross sections to be applied to more modern HAXPES energy ranges, which were crucially missing from previous cross section databases. Angular distribution parameters allow the user to investigate the effects of changing the polarisation of light on the outgoing photoelectron, particularly useful for probing metal s states at band edges. Data can be found in consistent Excel formatting, along with digitised versions of the Schofield, Yeh and Lindau datasets on figshare and on Dr. Anna Regoutz’ website. These are currently being implemented into the backend of GALORE.
Digitisation of Trzhaskovskaya Dirac-Fock Photoionisation Parameters for HAXPES Applications
If you do use GALORE, please cite the following paper in your publication:
A. J. Jackson, A. M. Ganose, A. Regoutz, R. G. Egdell and D. O. Scanlon, GALORE: Broadening and weighting for simulation of photoelectron spectroscopy, Journal of Open Source Software, 3, 773 (2018) DOI: 10.21105/joss.00773
Publications using GALORE
-
- K. T. Butler, G. S. Gautam and P. Canepa, Designing interfaces in energy materials applications with first-principles calculations, npj Computational Materials, 5, 19 (2019) doi: 10.1038/s41524-019-0160-9
-
- J. J. Bean and K. P. McKenna, Stability of point defects near MgO grain boundaries in FeCoB/MgO/FeCoB magnetic tunnel junctions, Physical Review Materials, 2, 125002 (2019) doi: 10.1103/PhysRevMaterials.2.125002
-
- A. Regoutz, A. M. Ganose, L. Blumentham, C. Schlueter, T.-L. Lee, G. Kielich, A. K. Cheetham, G. Kerherve, Y.-S. Huang, R.-S. Chen, G. M. Vinai, T. Pincelli, G, Panaccione, K. H. L. Zhang, R. G. Egdell, J. Lischner, D. O. Scanlon, and D. J. Payne, Insights into the Electronic Structure of OsO2 using Soft and Hard X-ray Photoelectron Spectroscopy in Combination with Density Functional Theory, Physical Review Materials, 3, 025001 (2019) doi: 10.1103/PhysRevMaterials.3.025001
Written by Dr John Buckeridge, CPLAP which stands for the Chemical Potential Limits Analysis Program (click here to get the source code), is a program designed to determine the thermodynamical stability of a material, and, if it is stable, to determine the ranges of the constituent elements’ chemical potentials within which it is stable, in comparison with competing phases and the elemental forms. CPLAP is extremely useful for Materials Design, as you can use it for testing the stability of new materials versus competing phases. It can also be used to set the boundaries of chemical potentials for defect Chemistry/Physics analysis (see figure below). For a full explanation, read the paper here.

If you do use CPLAP, please cite the following paper in your publication:
J. Buckeridge, D. O. Scanlon. A. Walsh and C. R. A. Catlow, Automated procedure to determine the thermodynamic stability of a material and the range of chemical potentials necessary for its formation relative to competing phases and compounds, Computer Physics Communications, 185(1), 330-338 (2014)
Publications using CPLAP
-
- Q. Chen, R. Zhang, J. Xu, S. Cao, Y. Guo, Y. Li, F. Gao, First-principles calculations of defect formation energy and carrier concentration of Ti4+, Ta5+ and W6+ doped KSr2Nb5O15, Computational Materials Science, Accepted (2018) doi: 10.1016/j.commatsci.2019.109427
-
- A. Moradabadi and P. Kaghazchi, Defect chemistry in cubic Li6.25Al0.25La3Zr2O12 solid electrolyte: A density functional theory study, Solid State Ionics, 338, 74 (2019) doi: 10.1016/j.ssi.2019.04.023
-
- J. Buckeridge, Equilibrium point defect and charge carrier concentrations in a material determined through calculation of the self-consistent Fermi energy, Computer Physics Communications, 244, 329 (2019) doi: 10.1016/j.cpc.2019.06.017
-
- A. Živković, A. Roldan, and N. H. de Leeuw, Tuning the electronic band gap of Cu2O via transition metal doping for improved photovoltaic applications, Physical Review Materials, 3, 115202 (2019) doi: 10.1103/PhysRevMaterials.3.115202
-
- H. Liu, Z. Yang, Q. Wang, X. Wang, and X. Shi, Atomistic insights into the screening and role of oxygen in enhancing the Li+ conductivity of Li7P3S11−xOx solid-state electrolytes, Physical Chemistry Chemical Physics, 21, 26358 (2019) doi: 10.1039/C9CP05329H
-
- Y.-K. Jung, J. Calbo, J.-S. Park, L. D. Whalley, S. Kim, and A. Walsh, Intrinsic doping limit and defect-assisted luminescence in Cs4PbBr6, Journal of Materials Chemistry A, 7, 20254 (2019) doi: 10.1039/C9TA06874K
-
- I. Elias, A. Soon, J. Huang, B. S. Haynes, and A. Montoya, Atomic order, electronic structure and thermodynamic stability of nickel aluminate, Physical Chemistry Chemical Physics, 21, 25952 (2019) doi: 10.1039/C9CP04325J
-
- S. Kim, J.-S. Park, S. N. Hood, and A. Walsh, Lone-pair effect on carrier capture in Cu2ZnSnS4 solar cells, Journal of Materials Chemistry A, 7, 2686 (2019) doi: 10.1039/C8TA10130B
-
- B. A. D. Williamson, G. J. Limburn, G. W. Watson, G. Hyett, and D. O. Scanlon, Computationally Driven Discovery of Layered Quinary Oxychalcogenides: Potential p-Type Transparent Conductors?, ? , Submitted (2019) ChemRxiv
-
- J. Buckeridge, T. D. Veal, C. R. A. Catlow, and D. O. Scanlon, Intrinsic disorder and the n- and -p-type dopability of the narrow band gap semiconductors GaSb and InSb, Physical Review B , 100, 035207 (2019) doi: 10.1103/PhysRevB.100.035207
-
- J. Buckeridge, C. R. A. Catlow, M. R. Farrow, A. J. Logsdail, D. O. Scanlon, T. W. Keal, P. Sherwood, S. M. Woodley, A. A. Sokol, and A. Walsh, The deep vs shallow nature of oxygen vacancies and consequent n-type carrier concentrations in transparent conducting oxides, Physical Review Materials , 2, 054604 (2018) doi: 10.1103/PhysRevMaterials.2.054604
-
- A. L. Galvin and G. W. Watson, Defects in orthorhombic LaMnO3 – ionic versus electronic compensation, Physical Chemistry Chemical Physics, 20, 19257 (2018) doi: 10.1039/C8CP02763C
-
- M. Rittiruam, A. Yangthaisong and T. Seetawan, Enhancing the Thermoelectric Performance of Self-Defect TiNiSn: A First-Principles Calculation, Journal of Electronic Materials, 47, 4456 (2018) doi: 10.1007/s11664-018-6686-7
-
- M. Quesada-Gonzalez, B. A. D. Williamson, C. Sotelo-Vasquez, A. Kafizas, N. D. Boscher, R. Quesada-Cabrera, D. O. Scanlon, C. J. Carmalt, and I. P. Parkin, A Deeper Understanding of Boron-doped Anatase Thin Films as a Multifunctional Layer through Theory and Experiment, Journal of Physical Chemistry C , 122, 714 (2018) doi: 10.1021/acs.jpcc.7b11142
-
- A. Walsh and A. Zunger, Instilling Defect Tolerance in New Compounds, Nature Materials, 16, 964 (2017) doi:10.1038/nmat4973
-
- A. L. Galvin and G. W. Watson, Modelling Oxygen Defects in Orthorhombic LaMnO3 and its Low Index Surfaces, Physical Chemistry Chemical Physics, 19, 24636 (2017) doi:10.1039/C7CP02905E
-
- Y. G. Yu, X, Zhang and A. Zunger, Natural Off-Stoichiometry Causes Carrier Doping in Half-Heusler Filled Tetrahedral Structures, Physical Review B, 95, 085201 (2017) doi:10.1103/PhysRevB.95.085201
-
- C. N. Savory, A. M. Ganose and D. O. Scanlon, Exploring the PbS-Bi2S3 Series For Next Generation Energy Conversion Materials, Chemistry of Materials, 29, 5156 (2017) doi: 10.1021/acs.chemmater.7b00628
-
- E. Olsson, X. Aparicio-Angles and N. H. de Leeuw, A Computational Study of the Electronic Properties, Ionic Conduction, and Thermal Expansion of Sm1−xAxCoO3 and Sm1−xAxCoO3−(x/2) (A = Ba2+, Ca2+, Sr2+, and x = 0.25, 0.5) as Intermediate Temperature SOFC Cathodes, Physical Chemistry Chemical Physics, 19, 13960 (2017) doi: 10.1039/C7CP01555K
-
- Z. Xie, Y. Sui, J. Buckeridge, C. R. A. Catlow, T. W. Keal, P. Sherwood, A. Walsh, D. O. Scanlon, S. M. Woodley, and A. A. Sokol, Demonstration of the donor characteristics of Si and O defects in GaN using hybrid QM/MM, Physica Status Solidi A, 214, 1600440 (2017) doi: 10.1002/pssa.201600445
-
- J. Kaczkowski and A. Jezierski, Effect of Chemical and Hydrostatic Pressure on Electronic Structure of BiPd2O4: A First-Principles Study, Journal of Alloys and Compounds, 726, 737 (2017) doi: 10.1016/j.jallcom.2017.08.030
-
- S. H. Shah and P. D. Bristowe, Point Defect Formation in M2AlC (M = Zr,Cr) MAX Phases and Their Tendency to Disorder and Amorphize, Scientific Reports, 7, 9667 (2017) doi: 10.1038/s41598-017-10273-6
-
- C. N. Savory, A. Walsh and D. O. Scanlon, Can Pb-free Halide Double Perovskites Support High-efficiency Solar Cells?, ACS Energy Letters , 1, 949 (2016) doi: 10.1021/acsenergylett.6b00471
-
- W. M. Linhart, M. K. Rajpalke, J. Buckeridge, P. A. E. Murgatroyd, J. J. Bomphrey, J. Alaria, C. R. A. Catlow, D. O. Scanlon, M. J. Ashwin and T. D. Veal, Band Gap Reduction in InSbxN1-x Alloys: Optical Absorption, k.P Modeling and Density Functional Theory , Applied Physics Letters, 109, 132104 (2016) doi: 10.1063/1.4963836
-
- M. R. Farow, C. R. A. Catlow, A. A Sokol and S. M. Woodley, Double Bubble Secondary Building Units Used as a Structural Motif for Enhanced Electron–hole Separation in Solids, Materials Science in Semiconductor Processing, 42, 147 (2016) doi: 10.1016/j.mssp.2015.08.023
-
- E. Olsson, X. Aparicio-Angles and N. H. de Leeuw, Ab Initio Study of Vacancy Formation in Cubic LaMnO3 and SmCoO3 as Cathode Materials in Solid Oxide Fuel Cells, The Journal of Chemical Physics, 145, 014703 (2017) doi: 10.1063/1.4954939
-
- F. H. Taylor, J. Buckeridge and C. R. A. Catlow, Defects and Oxide Ion Migration in the Solid Oxide Fuel Cell Cathode Material LaFeO3, Chemistry of Materials, 28, 8210 (2016) doi: 10.1021/acs.chemmater.6b03048
-
- J. Buckeridge, F. H. Taylor and C. R. A. Catlow, Efficient and Accurate Approach to Modeling the Microstructure and Defect Properties of LaCoO3, Physical Review B, 93, 155123 (2016) doi: 10.1103/PhysRevB.93.155123
-
- J. Buckerdige, D. Jevdokimovs, C. R. A. Catlow and A. A. Sokol, Nonstoichiometry and Weyl Fermionic Behavior in TaAs, Physical Review B, 94, 190101 (2016) doi: 10.1103/PhysRevB.94.180101
-
- J. Buckeridge, K. T. Butler, C. R. A. Catlow, A. J. Logsdail, D. O. Scanlon, S. A. Shevlin, A. A. Sokol, S. M. Woodley, and A. Walsh, Polymorph Engineering of TiO2: Demonstrating How Absolute Reference Potentials are Determined by Local Coordination, Chemistry of Materials, 27, 3844 (2015) doi: 10.1021/acs.chemmater.5b00230
-
- Z.-H. Cai, P. Narang, H. A. Atwater, S. Chen, C.-G. Duan, Z.-Q. Zhu and J.-H. Chu, Cation-Mutation Design of Quaternary Nitride Semiconductors Lattice-Matched to GaN, Chemistry of Materials, 7757, (2015) doi: 10.1021/acs.chemmater.5b03536
-
- R. D. Bayliss, S. N. Cook, D. O. Scanlon, S. Fearn, J. Cabana, C. Greaves, J. A. Kilner and S. J. Skinner, Understanding the Defect Chemistry of Alkali Metal Strontium Silicate Solid Solutions: Insights from Experiment and Theory, Journal of Materials Chemistry A, 2, 17919 (2014) doi: 10.1039/c4ta04299a
-
- D. O. Scanlon, J. Buckeridge, C. R. A. Catlow, G. W. Watson, Understanding doping anomalies in degenerate p-type semiconductor LaCuOSe, Journal of Materials Chemistry C, 2, 3429 (2014) doi: 10.1039/c4tc00096j
-
- C. Wang, S. Chen, J.-H. Yang, L. Liang, H.-J. Xiang, X.-G. Gong, A. Walsh and S.-H. Wei, Design of I2–II–IV–VI4 Semiconductors through Element Substitution: The Thermodynamic Stability Limit and Chemical Trend, Chemistry of Materials, 26, 3411 (2014) doi: 10.1021/cm500598x